Chemistry Discussion Thread
- Thread starter MrEDuck
- Start date
MrEDuck
Well-Known Member
Sonar, I don't know very much about the production of sassafras oil. I know it's made by steaming the roots of a sassafras tree. No idea about the yield. I assume one tree should give a decent amount. I also assume it's a fuckload of work. Chopping a tree is one thing, digging up a few feet of roots is another. You'd need to figure out what kinda price you can get for bulk MDMA and do a cost benefit analysis. If you're doing it for headies, then I admire your work ethic.
MrEDuck
Well-Known Member
Because I just unpacked my copy, if you are going to ever try any experiments the one chemistry book you must buy is The Organic Chem Lab Survival Manual by James Zubrick. It's probably on its 5th or 6th edition now, any edition is fine. I can't believe I forgot to mention it earlier.
MrEDuck
Well-Known Member
The synthesis of phenethylamines and amphetamines via the Knoevengal Condensation. The Knoevengel condensation is great for making nitrostyrenes from benzaldehydes and nitromethane, or whatever you would call the amphetamine equivalent of a nitrostyrene by using nitroethane instead. These can then be easily reduced to phenethylamines or ampetamines by using reducing agents like lithium aluminum hydride or an Al/Hg amalgam. Most of the syntheses in PiHKAL use this reaction because it is an easy reaction with good yields. It can be run with a few different reaction conditions, solvent free using the nitro(m)ethane as both solvent and reagent in the presence of ammonium acetate as a catalyst, using acetic acid as the solvent and keeping the ammonium acetate, or my favorite way in the microwave using the nitro(m)ethane as solvent and base treated silica beads as the catalyst. The first two usually take 2hr-24hr of heating on the steambath to achieve good yields. I once discovered a bottle of 3,4,5 trimethoxybenzaldehyde looking for something else and decided to try the MW reaction. 5 mins and I had 88% yield of the nitrostyrene. I had several grams of mescaline within hours! Needless to say I was sold on microwave chemistry. One of the very interesting properties of a microwave is that the boiling points of many solvents are higher in a microwave than when they are heated thermally. So it's like using a pressure cooker to cook something faster because of boiling point elevation.
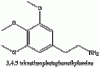
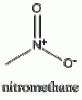
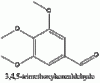
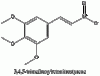
Above we have images of the various structures in the synthesis of mescaline (chem draw wasn't pulling its structure from mescaline so I just used it's IUPAC name). The nitromethane and the aldehyde bond losing a molecule of water (any condensation reaction is one where new bonds are formed and a molecule of water is lost) to form the betanitrostyrene, which is reduced to mescaline. If anyone wants me to draw out the mechanism I will.
I'm making progress on getting the LSD post done. There's so many fucking structures to draw though. I'll only have net access through my phone for the next day or so. Hopefully I can finish it up for Friday, otherwise it'll probably have to wait until Monday or Tuesday.
Because I was asked about 2Cs specifically as well I'll go over that quickly here. Generally they start in one of two ways. For 2C-H and the halogenated ones you start with 2,5 dimethoxybenzaldehyde, condense with nitromethane, and reduce to 2C-H. You can then tack on halogen by various methods. Like reacting it with elemental bromine to make 2C-B (why would you make another 2C? we know B is the king of the class). For the alkylated ones like -D and -E the starting material is p-dimethoxybenzene, which is alkylated with a Friedel-Crafts alkylation, then methylated and oxidized to an aldehyde. I imagine the purification for -D is a bitch. But again if you're making 2Cs why are you trying to make something that isn't B? I'll draw the 2 general syntheses out after I get done with the LSD post.
Above we have images of the various structures in the synthesis of mescaline (chem draw wasn't pulling its structure from mescaline so I just used it's IUPAC name). The nitromethane and the aldehyde bond losing a molecule of water (any condensation reaction is one where new bonds are formed and a molecule of water is lost) to form the betanitrostyrene, which is reduced to mescaline. If anyone wants me to draw out the mechanism I will.
I'm making progress on getting the LSD post done. There's so many fucking structures to draw though. I'll only have net access through my phone for the next day or so. Hopefully I can finish it up for Friday, otherwise it'll probably have to wait until Monday or Tuesday.
Because I was asked about 2Cs specifically as well I'll go over that quickly here. Generally they start in one of two ways. For 2C-H and the halogenated ones you start with 2,5 dimethoxybenzaldehyde, condense with nitromethane, and reduce to 2C-H. You can then tack on halogen by various methods. Like reacting it with elemental bromine to make 2C-B (why would you make another 2C? we know B is the king of the class). For the alkylated ones like -D and -E the starting material is p-dimethoxybenzene, which is alkylated with a Friedel-Crafts alkylation, then methylated and oxidized to an aldehyde. I imagine the purification for -D is a bitch. But again if you're making 2Cs why are you trying to make something that isn't B? I'll draw the 2 general syntheses out after I get done with the LSD post.
MrEDuck
Well-Known Member
Canndo you can determine safrole content of sassafras oil by it's congealing point. There's a paper in the Rhodium archive http://www.erowid.org/archive/rhodium/chemistry/safrole.congealing.html
There's no way to know until you have it in hand though.
There's no way to know until you have it in hand though.
MrEDuck
Well-Known Member
I hope you won't just be lurking if you have knowledge. What are you planning to specialize in now that you've graduated?looks like ill be lurking on this thread, chemistry junkie who just graduated, havent done much drug synths but knowledgeable on all processes, theories, setups, structures etc...
fourtwentychat
Well-Known Member
Lets suppose that you were given a half-cup of concentrated cactus tea - a couple feet worth of Bridgesii, for example. Assuming that you would be unable to drink the tea as-is, what would be the simplest way for you to proceed, while using only the very minimum in terms of Chemistry tools and materials?
ndangerspecimen101
Well-Known Member
How about Nutmeg or Black Seed Oil. Certainly fractional distillation will have to take place before anything else gets started.Some notes about MDMA synthesis. Let's start with Bright Star. It's a workable synth, but it does have it's faults. First would be the use of peanut oil. Get a real high temp silicone oil. No one watches them and they don't smoke and don't have a habit of splattering (200°C+ oil hurts like a mofo, or hellfire as a research advisor of mine said). Next up is the methylamine from formaldehyde and ammonia. Jesus fucking Christ do not use this procedure as written. When it gets to the point where you start pulling vacuum turn the heat off and let it cool a minimum of 50°C. Never turn vacuum on a hot distillation no matter how careful you think you are. I used to do analytical distillations when I worked in alt fuel petrochemical research. The first fraction was done at atmospheric pressure, and the. You cooled it before turning on the vacuum, at least that's what you do if you don't want to see the mother of all bbumps, ruin your distillation and make one hell of a mess.
Next what's up with using DCM as a solvent? Yuck, find a substitute. Also look into alternatives to DMF for the Wacker oxidation. That's some toxic shit. In the distillation don't use vacuum for the first fraction. Which should not be DCM because don't use halogenated organics as solvents if you can avoid it. Finally I don't care if you are Woodward himself recrystallize that shit. It was just sitting in a solution of mercury. If you don't recrystallize it and give it to another person you deserve to be shot in the genitals.
So let's look at dr Drool's synth. This also starts with Safrole and uses DCM. Apparently the Hive died because everyone got cancer (seriously halogenated organics are bad). Why would someone vacuum distill nitromethane? You're gonna lose half of it to your poor pumps oil. If something boils at <150°C do an atmospheric distillation.
OMG a ziplock bag has no place around chemicals. Rh where's the note saying don't do this. The reductive ammination looks sound. Do these guys have something against recrystallization? Does that ziplock bag give it some extra kick.
So both synths are reasonable, provided some changes are made for the sake of safety (sanity?). Except that safrole is harder to get your hands on than a pig in a lube factory. So how do we get around this? We can make it ourselves of course. Back in the late nineties bulk sassafras oil was getting more difficult to obtain. Needless to say this was a major problem for some people. Well it turns out that if you heat allylcatechol sufficiently (~250°C IIRC) it thermally rearranges to to 1-allyl-3,4dihydroxybenzene. That's a methylene bridge away from being safrole. Hydroxy groups are pretty good nucleophiles, and deprotonation only makes em better. So it turns out dihalomethanes have a use in MDMA synth. Just as reagents, not solvents. DBM or DIM (Cl is a crap leaving group) mixed with your allylcatechol and some base and wala, you have safrole.
Sorry it took me a few days to get this posted. I've felt terrible the last few days
ndangerspecimen101
Well-Known Member
It has be done. With great success. There's a heap load of work to be done before that sassafras is turned into safrole.Maybe a silly question but would it be possible to extract a usable amount of safrole from sassafras plant material? The stuff grows everywhere around here.
Luckily, sassafras is the most highest yielding compound of safrole, without other alkaloid derivatives that will necessarily effect the end product.
ndangerspecimen101
Well-Known Member
Some don't contain safrole due to FDA restriction on things containing safrole. As safrole is known to be carcinogenic.As an aside now but I have to ask:
I have read that some sassafras oil does not contain safrole - how can we tell? (there is oil for sale on e-bay)
Root Beer and other products use other means of flavoring besides safrole now.
MrEDuck
Well-Known Member
It'd be just fine Canndo. It might have degraded a bit but there should be plenty of useful safrole left.
To the guy with the gross cactus tea you need to do an acid base extraction. I wrote out a how to but the Internet ate it apparently. I'll rewrite it ASAP. Hope everyone's days are going better than mine. Talk to y'all soon.
To the guy with the gross cactus tea you need to do an acid base extraction. I wrote out a how to but the Internet ate it apparently. I'll rewrite it ASAP. Hope everyone's days are going better than mine. Talk to y'all soon.
ndangerspecimen101
Well-Known Member
How many grams of safrole might be converted from (1) liter of sassafras MrEDuck?
In addition, I have a few questions for you that I'll address in PM.
In addition, I have a few questions for you that I'll address in PM.
canndo
Well-Known Member
How many grams of safrole might be converted from (1) liter of sassafras MrEDuck?
In addition, I have a few questions for you that I'll address in PM.
TOOK the words right off of my keyboard Ndanger. (given that the person knew exactly how do execute the proceedures so that there was little waste - of course)
morfin56
New Member
Wouldn't that have to many factors to account for?How many grams of safrole might be converted from (1) liter of sassafras MrEDuck?
In addition, I have a few questions for you that I'll address in PM.
You always have really fine looking sand, makes me wonder.. LOL I'm not saying anything that you PROBABLY already KNOW.
ndangerspecimen101
Well-Known Member
Exactly.Wouldn't that have to many factors to account for?
You always have really fine looking sand, makes me wonder.. LOL I'm not saying anything that you PROBABLY already KNOW.
Rough estimate.
Each tree specimen is different. And the procedures involved in a distillation (Fractional Distillation) can vary to person to person. But, never hurts to formulate an educated guess.
MrEDuck
Well-Known Member
Fourtwentychat, I don't blame you for not wanting to consume that. Cactus tea is gross. I'd use an acid base extraction on it. Start by acidifying the tea to pH 2-3 to make good and sure everything is ionized. Then wash with a nonpolar solvent. D-limonene is nice because food grade is readily available and inexpensive. Separate (a 500mL sep funnel is available from American Science and Surplus, sciplus.com, for $26) and pitch the organic (nonpolar) layer. It has all the fats and waxes that need to be removed to get a decent product instead of a gooey mess. Now raise the pH to 11.5-12, you should see a milky precipitate form. Now wash with nonpolar solvent and separate, save the nonpolar layer and wash the aqueous layer 3x with ~25mL of nonpolar solvent, save the aqueous layer for the moment in case something went wrong, it can be discarded once you have product. Pool the organic layers and wash them 3x with saturated NaCl solution (about 25mL per wash). Now you need to put some acid in to salt out the mescaline. I like dry HCl but that's a technique you shouldn't use if you aren't very familiar with labwork and know how to handle it, breathing that shit in is awful.
There's plenty of teks on the net. If you can concentrate vinegar (done by steadily adding ethyl acetate to the vinegar to form a low boiling azeotrope with the water, then distilling the remaining acetic acid once all the water and ethyl acetate have been boiled off) glacial acetic acid is awesome, you just pour it into the nonpolar layer and watching the crystals fall out of solution.
Everyone asking about safrole content of sassafras oil, the concentration varies a lot, but good sass oil should be in the range of 50-70% safrole. Safrole is slightly denser than water (~1.1g/mL) so if you can get 90% of it out you'll get about 45-65% of the original volume, which means about 500-650g of safrole. And you can get about 50% of that weight as product (varies based on method and experience).
There's plenty of teks on the net. If you can concentrate vinegar (done by steadily adding ethyl acetate to the vinegar to form a low boiling azeotrope with the water, then distilling the remaining acetic acid once all the water and ethyl acetate have been boiled off) glacial acetic acid is awesome, you just pour it into the nonpolar layer and watching the crystals fall out of solution.
Everyone asking about safrole content of sassafras oil, the concentration varies a lot, but good sass oil should be in the range of 50-70% safrole. Safrole is slightly denser than water (~1.1g/mL) so if you can get 90% of it out you'll get about 45-65% of the original volume, which means about 500-650g of safrole. And you can get about 50% of that weight as product (varies based on method and experience).